Related: how to download bioinformatics data example - FASTq files from scRNA-seq
- go to 10x genomics/go to the 10x downloads website
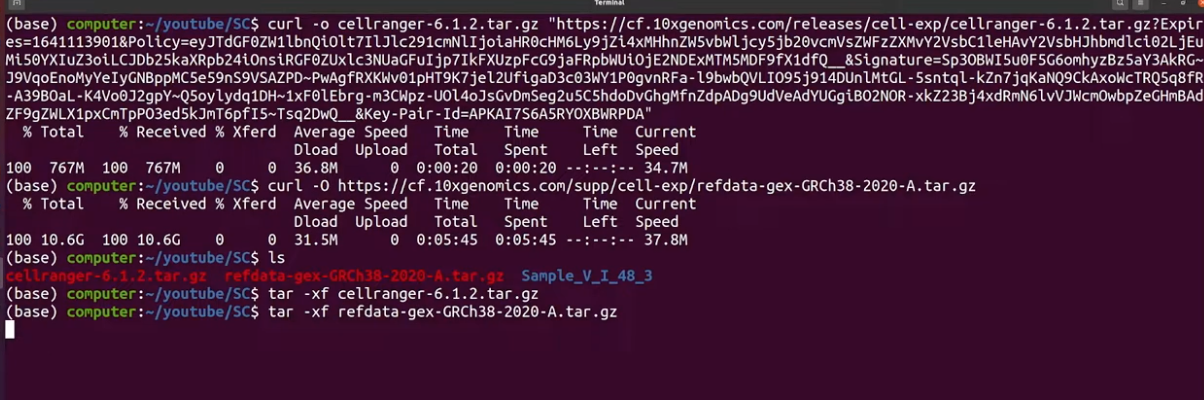
- copy the cellranger binary for either curl or wget and paste it in your terminal
- go back to the downloads page and get the references that you need, paste it into the terminal
- if the data is from humans, you’d get the human references; from mice, the mice dataset, etc.
- another organism = need to build your own custom 10x
- open the cellranger file and then open the ref-file as well
- you can free up space by using
rm *.gzto remove all gz files (which are similar to ZIP files) i.e. compressed files
- use
lsto make sure you have those files downloaded properly - verify file names, the nomenclature has to be specific otherwise it won’t work correctly
- the highlighted section below are the read names from the files, in this section it comes off of an illumina sequencer
- if you’re getting it from an open database or somewhere else, there’s a chance that your file names are different so you’ll either have to look for the original file names or just guess…

- you need to point to the actual binary for cellranger in order for it to work. then need to point it to the downloaded data, then the ref data
cellranger-7.2(V. number etc.)/cellranger count --id tutorial_sample --transcriptome refdata-gex-GRCh38-2020-A/ --fastqs Sample_V_I_48_3 --(sample prefix) --expect-cell 10000 --localcore 20 --localmem 100 - localcore and localmem - the higher the numbers, the less time it will take. depends on your computer specs
- even with this a sample can take up to 1 hour per sample
- transcriptome is the reference directory
- fastqa is the directory containg the FASTA and FASTQ
- expect-cells will show you around the amount of cells present when you did the wetlab experiment. never an exact number, aim for ballpark for when they formed the single-cell suspension (10,000)
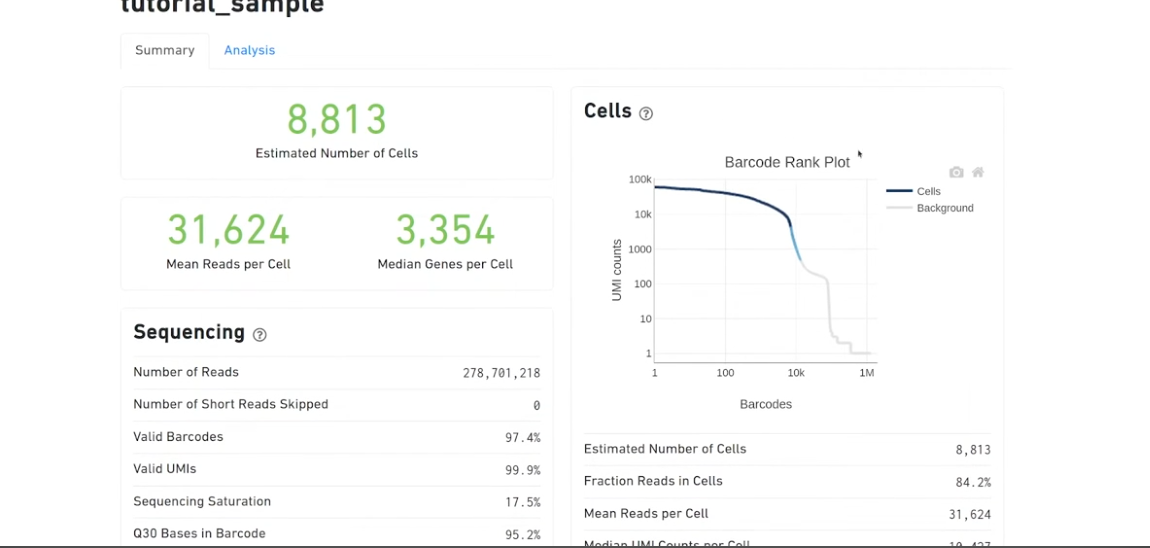
- once it’s done, can check the
web_summary.html- if you ran it with a remote server, you’d have to download the files
- whatever
--iddirectory you’ve created (in this example it istutorial_sample), is the one where all the outputs are going to be stored in
firefox tutorial_sample/outs/web_summary.html